Opportunities and Challenges in the Digitalization of Production Control for Medical Products and Pharmaceuticals

The current discussions on the availability of drugs, for example in the case of flu medicines, antibiotics and cancer drugs, show just how important medical products and especially pharmaceuticals are. There are various approaches that are being taken to reinforcing supply security and counteracting bottlenecks – including laws designed to reduce supply bottlenecks for patent-free medicines and to improve the supply of medicines for children1. A topic that keeps cropping up is the need to strengthen production in Europe and to optimize the regulatory requirements for manufacturing and approval.
Right now, however, digitalizing production control using a line controller, for example, can help manufacturers get ahead in maximizing their production capacities and preventing possible supply bottlenecks in the process. Assuming the line controller is GMP-compliant, manufacturers can also benefit from the digitalization of the extensive requirements – from integration to reporting. This represents a major step towards ensuring quality-relevant processes in terms of product liability, certifications and traceability.
In our interview, we ask Daniel Hahm, our business analyst responsible for factory automation, what opportunities and challenges arise as a result. <Bild Daniel>
Daniel, When We Refer to Medical Products and Pharmaceuticals, What Exactly Do We Mean Here?
Pharmaceuticals are substances, drugs and medicines that are used to diagnose, treat or prevent diseases, so this means products such as tablets and vaccines, for example. Medical products, such as implants or syringes, are used for therapeutic or diagnostic purposes. The German Federal Institute for Drugs and Medical Devices (BfArM) defines medical devices as “products with a medical purpose that are intended by the manufacturer for use in humans. In contrast to drugs that have pharmacological, immunological or metabolic effects, the intended main effect of medical devices is primarily achieved, for example, by physical means.” 2 The main focus is therefore on the intended purpose – a reason why there is a separate regulatory framework for this segment.
Which Standards and Directives Do Manufacturers of Medical Products Need to Comply with When Producing in the EU?
The standards that are decisive for the production and approval of medical products are the European Medical Product Regulation 2017/745 (MDR) and the EU In-Vitro Diagnostics Regulation 2017/746 (IVDR). In Germany, the medical product act (MPDG) applies at national level to implement and supplement these regulations. These three together regulate the marketing and commissioning of medical products in order to ensure the safety, suitability and performance of medical products as well as the health and protection of patients, users and third parties.
The industry-specific process and quality standard DIN EN ISO 13485 “Medical Products: Quality Management Systems – Requirements for Regulatory Purposes” still applies to the design and manufacture of medical products. It is the only harmonized standard for the EU MDR and IVDR regulations and consequently represents the industry standard for demonstrating compliance with the requirements of the EU regulations for a comprehensive quality management system. Although it is not obligatory in the EU, the vast majority of medical product manufacturers cannot get around providing appropriate certification and implementation.
And What about Pharmaceuticals?
These also require approval for manufacturing (manufacturing permit) and marketing (approval). In order to ensure the quality, effectiveness and safety of drugs and active ingredients, measures need to be taken to ensure the required quality during production. These quality assurance requirements for production processes and the shopfloor are formulated in the principles and guidelines of Good Manufacturing Practice (GMP) for pharmaceutical products for human use (EU GMP Directive 2017/1572). The interpretation of these GMP principles is described in the EU GMP guidelines. The GMP requirements it defines must be complied with by the pharmaceutical industry in the EU and they are a mandatory requirement for the granting and maintenance of a manufacturing permit by the relevant supervisory authorities. In Germany, the application of good manufacturing practice has been integrated into national law by the Medicines Act (AMG) and the Medicines and Active Ingredient Manufacturing Regulation (AMWHV).
There are also additional GMP guidelines formulated, for example, by the Pharmaceutical Inspection Co-Operation Scheme (PIC/S), the World Health Organization (WHO) and by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH). Although these directives and guidelines are not legally binding, they have become established as standard regulations in the pharmaceutical industry.
GMP or GAMP – What Is the Difference?
GAMP stands for Good Automated Manufacturing Practice in relation to computer-aided systems. The International Society of Pharmaceutical Engineering (ISPE) has published a guideline called “GAMP 5: A risk-based approach for compliant GxP computer-aided systems”, which, despite the lack of legal obligation, is generally recognized among manufacturers and suppliers as a standard set of rules for validating computer-aided systems in the pharmaceutical industry. The reason for this is that the GAMP guidelines take a consistent risk-based approach, which is in line with Annex 11 of the EU GMP guideline for computer-aided systems and contains pragmatic guidelines, approaches and practical tools. In addition to the frequently applied V model for linear software development, it also describes a wide range of other approved validation approaches, process models, development methods and scenarios, including agile software development tactics such as SCRUM.
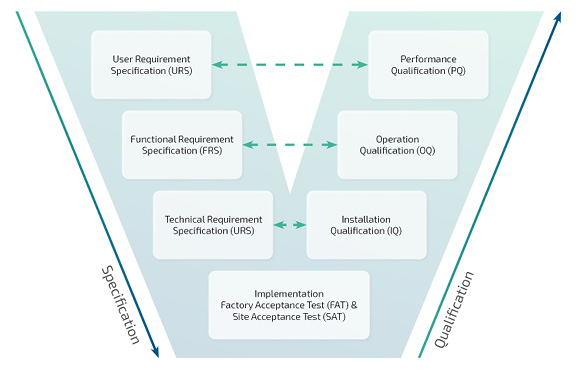
In Modern Factories, Production Control Systems Handle All the Manufacturing Processes. What Does a Production Control System Do in the Specific Area of Manufacturing Medical Products and Pharmaceuticals?
A production control system (or line controller)can process work tasks and steps autonomously, or with the intervention of operators through user interfaces. The strategy chosen depends on the specific production process in each case. For example, a line controller can handle different roles such as
- recipe, user and production equipment management for a Manufacturing Execution System (MES),
- step-by-step, documented manufacturing processes for a Quality Assurance (QA) system, or
- batch documentation review at the end of the production process.
How Is the Validation Process Carried out for a Line Controller That Needs to Be GMP-Compliant?
There is no general answer to this question, as each line controller is usually specifically adapted and customized to meet the manufacturer’s requirements. According to Annex 11 of the EU GMP guideline “decisions on the extent of validation and data integrity controls should be based on a justified and documented risk assessment of the computerized system“, and “The validation documentation and reports should cover all the life-cycle stages of the system”3. As a result, the scope and documentation of the validation of a computer-aided system must generally be determined on a risk basis and depends on the selected development process and thus on the life cycle of the system. There can therefore be no standard procedure for validating a line controller.
What Should Pharmaceutical Manufacturers Consider in Advance if a Line Controller in a Production Plant Is to Be Upgraded or Replaced?
First of all, the technical solution should not be defined or specified in advance but should be based on the challenges specific to the respective production process. Important preliminary considerations for production control include:
- Which corporate, project or product goals should be achieved with the proposed but not yet available solution?
For example, it could be a matter of simplifying work steps, shortening processing times, eliminating paper, or introducing the automatic recording of quality attributes. Ideally, the current situation and the desired target situation after the solution has been launched are described before the proposed solution is introduced.
- What is the usage context of the future system?
Without an understanding of the underlying usage context of the future system, it is hardly possible to develop a system adequately. Business processes and sub-processes (sequences of individual activities, which are to be carried out step-by-step manually/automatically in order to achieve a technical goal) must therefore be described in detail and modelled if necessary to outline the resulting scenarios or applications for the system.
What Belongs to the System and What Belongs to the System Context?
This question should be answered as early as possible, ideally before documenting the actual requirements in a specification, in order to decide who and what has influence on the requirements of a system (sources of requirements) and to know the scope of the system and system development (in-scope vs. out-of-scope). Only with a fully defined system context is it possible to fully understand the system requirements and incorporate them into system development. Here, the system stands for the computer-aided system (e.g. a line controller with software (modules), hardware components, system documentation), the system context for the relevant environment (e.g. the production plant, employees, other software systems) and the system boundary for the interfaces between them (e.g. data that employees should be able to enter into the system). Defining the system context is crucial for answering three important questions:
- What should be developed (customized and modified)?
- What has an influence on development (not customized and modified, but must be considered when determining system requirements)?
- What can be neglected (according to current knowledge)?
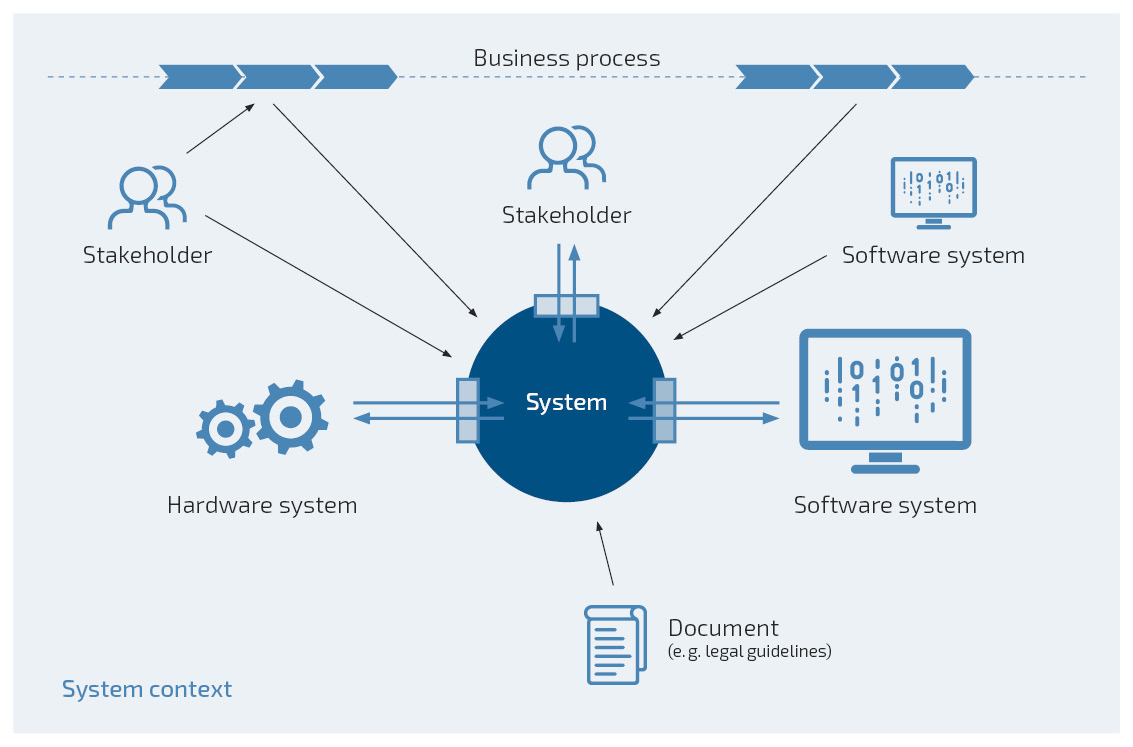
Who Are the Stakeholders of the Future System, What Needs and What Influence Do They Have on the Solution to Be Developed?
This should take into account all internal and external stakeholders who have direct or indirect demands on, or interests in, the solution to be developed (e.g. production control). This includes, for example, the people and groups who will operate, maintain or train the future system.
How Can Kontron AIS Help Digitalize Production?
Because we are the developer and integrator of software solutions for production control and connectivity in an GMP environment, customers can benefit from proven products such as our FabEagle®LC line controller and our FabEagle®Connect low-code interface integration software. Both our products and project development meet GMP requirements and are based on GAMP guidelines to ensure smooth integration according to the V model. These form the basis from which a variety of customized and process-specific extensions can also be implemented.
In addition to the appropriate software, systematic requirements engineering is crucial for the sustainable digitalization of analog production processes. That is why we assist with the identification, documentation, analysis and coordination of customer requirements and system specifications to be defined in the specification documents as well as requirements management over the life cycle. In workshops, we work together with the customer to define the specific requirements of the production process in terms of production control and connectivity.
1 https://www.recht.bund.de/bgbl/1/2023/197/VO.html, 31.08.2023
2 https://www.bfarm.de/DE/Medizinprodukte/_node.html; 30.08.2023
Do You Need Help Digitalizing Your Production in Compliance with GMP and GAMP Guidelines? I Am Your Partner to Talk To!
