Chancen und Herausforderungen der Digitalisierung der Produktionssteuerung von Medizinprodukten und Pharmaka

Wie wichtig Medizinprodukte und vor allem Pharmaka sind, zeigen die aktuellen Diskussionen rund um das Thema der Arzneimittelverfügbarkeit zum Beispiel bei Fiebersäften, Antibiotika oder Krebsmedikamenten. Um die Versorgungssicherheit zu stärken und Lieferengpässen entgegenzuwirken, werden verschiedene Ansätze herangezogen – auch entsprechende Gesetze wie das zur Bekämpfung von Lieferengpässen bei patentfreien Arzneimitteln und zur Verbesserung der Versorgung mit Kinderarzneimitteln1. Dabei immer wieder in der Diskussion: die Stärkung der Produktion am Standort Europa und die Optimierung der regulatorischen Anforderungen für die Herstellung und Zulassung.
Eine Digitalisierung der Produktionssteuerung, zum Beispiel durch ein Leitsystem, kann Hersteller jedoch auch schon heute dabei helfen, ihre Produktionskapazitäten zu maximieren und damit möglichen Lieferengpässen vorzubeugen. Ist ein solches Leitsystem noch GMP-konform, profitieren die Anlagenbetreiber zusätzlich von der Digitalisierung der umfangreichen Anforderungen – von der Integration bis zum Reporting. Damit ist ein großer Schritt in Richtung Absicherung qualitätsrelevanter Prozesse hinsichtlich Produkthaftung, Zertifizierungen und Rückverfolgbarkeit getan.
Was es dabei alles zu beachten gilt und welche Chancen, aber auch Herausforderungen sich daraus ergeben, fragen wir im Interview Daniel Hahm, der bei uns als Business Analyst den Bereich Fabrikautomation betreut.
Daniel, wenn wir von Medizinprodukten und Pharmaka sprechen, von was reden wir da eigentlich genau?
Unter Pharmaka werden Wirkstoffe, Arzneistoffe und Arzneimittel verstanden, die zur Diagnose, Therapie oder Prophylaxe von Krankheiten eingesetzt werden, also zum Beispiel Tabletten oder Impfstoffe. Medizinprodukte, wie zum Beispiel Implantate oder Spritzen, werden zu therapeutischen oder diagnostischen Zwecken verwendet. Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) definiert Medizinprodukte als „Produkte mit medizinischer Zweckbestimmung, die vom Hersteller für die Anwendung beim Menschen bestimmt sind. Anders als bei Arzneimitteln, die pharmakologisch, immunologisch oder metabolisch wirken, wird die bestimmungsgemäße Hauptwirkung bei Medizinprodukten primär auf z. B. physikalischem Weg erreicht.“ 2 Der Hauptunterschrift liegt also in der in der bestimmungsgemäßen Hauptwirkung, der sogenannten Zweckbestimmung – ein Grund, weswegen für beides auch unterschiedliche regulatorische Rahmen gelten.
Welche Normen und Richtlinien müssen die Hersteller von Medizinprodukten denn bei der Produktion in der EU beachten?
Bei den Medizinprodukten sind für die Herstellung und Zulassung die europäische Medizinprodukteverordnung 2017/745 (Medical Device Regulation, MDR) und die EU-Verordnung für In-vitro-Diagnostika 2017/746 (In-vitro Diagnostics Regulation, IVDR) maßgeblich. Zur Durchführung und Ergänzung dieser Verordnungen greift auf nationaler Ebene das Medizinprodukterecht-Durchführungsgesetz (MPDG). Diese drei zusammen regeln das Inverkehrbringen und die Inbetriebnahme von Medizinprodukten, um die Sicherheit, Eignung und Leistung der Medizinprodukte sowie die Gesundheit und den erforderlichen Schutz von Patient*innen, Anwender*innen und Dritten sicherzustellen.
Für das Design und die Herstellung von Medizinprodukten ist weiterhin die branchenspezifische Prozess- und Qualitätsnorm DIN EN ISO 13485 „Medizinprodukte: Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke“ relevant. Sie ist die einzige harmonierte Norm für die EU-Verordnungen MDR und IVDR und stellt damit den De-facto-Branchenstandard für den Nachweis der Erfüllung der Anforderungen der EU-Verordnungen an ein umfassendes Qualitätsmanagementsystem dar. Obwohl sie in der EU nicht obligatorisch ist, kommen also die allermeisten Medizinproduktehersteller um eine entsprechende Zertifizierung und Implementierung nicht herum.
Und wie sieht es bei den Pharmaka aus?
Auch diese bedürfen einer Genehmigung für das Herstellen (Herstellungserlaubnis) und das Inverkehrbringen (Zulassung). Um die Qualität, Wirksamkeit und Unbedenklichkeit von Arzneimitteln und Wirkstoffen sicherzustellen, müssen Maßnahmen getroffen werden, um die Herstellung in der vorgeschriebenen Qualität abzusichern. Diese Anforderungen an die Qualitätssicherung der Produktionsabläufe und -umgebung sind in den Grundsätzen und Leitlinien der Guten Herstellungspraxis (Good Manufacturing Practice, GMP) für Humanarzneimittel (EU-GMP-Richtlinie 2017/1572) formuliert. Die Auslegung dieser GMP-Grundsätze ist im EU-GMP-Leitfaden beschrieben und die darin definierten GMP-Anforderungen sind von der pharmazeutischen Industrie in der EU verbindlich einzuhalten und für die Erteilung und Aufrechterhaltung einer Herstellungserlaubnis durch die zuständigen Aufsichtsbehörden zwingende Voraussetzung. In Deutschland ist die Anwendung der Guten Herstellungspraxis durch das Arzneimittelgesetz (AMG) und die Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) in nationales Recht umgesetzt.
Des Weiteren gibt es ergänzende GMP-Richtlinien, die zum Beispiel durch das Pharmaceutical Inspection Co-Operation Scheme (PIC/S), die WHO (World Health Organization) oder durch das International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) erstellt werden. Obwohl diese Richtlinien und Leitfäden nicht rechtsverbindlich sind, haben sie sich als Standardregelwerke in der pharmazeutischen Industrie etabliert.
GMP oder GAMP – wo liegen die Unterschiede?
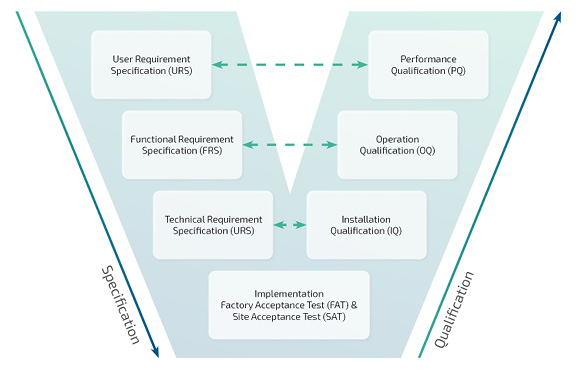
GAMP steht für Good Automated Manufacturing Practice, also eine gute automatisierte Herstellungspraxis, die sich auf computergestützte Systeme bezieht. Die International Society of Pharmaceutical Engineering (ISPE) hat unter der Bezeichnung „GAMP 5: Ein risikobasierter Ansatz für konforme GxP-computergestützte Systeme“ einen Leitfaden veröffentlicht, der sich trotz fehlender gesetzlicher Verbindlichkeit als allgemein anerkanntes Standardregelwerk für die Validierung computergestützter Systeme in der pharmazeutischen Industrie bei Herstellern und Zulieferern etabliert hat. Der Grund dafür: der GAMP-Leitfaden verfolgt einen konsequent risikobasierten Ansatz, der im Einklang mit Anhang 11 des EU-GMP-Leitfadens für computergestützte Systeme steht und pragmatische Leitlinien, Ansätze und Werkzeuge für die Praxis enthält. So ist neben dem häufig angewandten sogenannten „V-Modell“ zur linearen Softwareentwicklung auch das breite Spektrum anderer zugelassener Validierungsansätze, Vorgehensmodelle, Entwicklungsmethoden und -schemen beschrieben, einschließlich agiler Softwareentwicklungsmethoden wie SCRUM.
In modernen Fabriken übernehmen Produktionsleitsysteme die Steuerung der gesamten Fertigungsprozesse. Welche Aufgaben hat so ein Leitsystem in dem speziellen Bereich der Herstellung von Medizinprodukten und Pharmaka?
Ein Leitsystem kann Arbeitsaufgaben und -schritte selbstständig oder mithilfe des Eingriffs von Bediener*innen über sogenannte Benutzerschnittstellen übernehmen. Die Aufteilung zwischen beiden hängt stark von den individuellen Produktionsprozessen ab. So kann ein Prozessleitsystem (Leitrechner) zur Liniensteuerung zum Beispiel Elemente
- eines Produktionsleitsystems (MES) wie Rezept-, Nutzer- und Produktionsmittelverwaltung enthalten,
- Aspekte eines Qualitätssicherungssystems unterstützen, zum Beispiel durch schrittweise geführte und dokumentierte Herstellungsprozesse oder
- die Möglichkeit zur Überprüfung der Chargendokumentation am Ende des Produktionsprozesses bieten.
Wie erfolgt der Validierungsprozess eines Leitrechners zur Produktionssteuerung einer Fertigungslinie mit GMP-Anforderungen?
Darauf lässt sich keine pauschale Antwort geben, da die Leitrechner in der Regel für die entsprechenden Hersteller spezifisch angepasste und maßgeschneiderte Produktionssteuerungssysteme sind. Entsprechend Anhang 11 des EU-GMP-Leitfadens „… sollten Entscheidungen über den Umfang der Validierung und die Sicherstellung der Datenintegrität auf einer begründeten und dokumentierten Risikobewertung des computergestützten Systems basieren“ und „Die Validierungsdokumentation und -berichte sollten die maßgeblichen Phasen des Lebenszyklus abbilden“3. Daraus ergibt sich, dass der Umfang und die Dokumentation der Validierung eines computergestützten Systems generell risikobasiert ermittelt werden muss und vom gewählten Entwicklungsprozess und damit vom Lebenszyklus des Systems abhängig ist. Es kann daher kein Standardverfahren zur Validierung eines Leitrechners geben.
Was sollten pharmazeutische Hersteller im Vorfeld beachten, wenn ein Leitrechner in einer Produktionsanlage aufgerüstet oder ersetzt werden soll?
Zunächst einmal sollte die technische Lösung nicht vorab definiert oder vorgegeben werden, sondern sich an den Herausforderungen des jeweiligen individuellen Produktionsprozesses orientieren. Wichtige Überlegungen für eine Produktionssteuerung sind im Vorfeld dazu:
- Welche Unternehmens-, Projekt- oder Produktziele mit der angedachten, aber noch nicht vorhandenen Lösung erreicht werden sollen?
Es kann zum Beispiel darum gehen Arbeitsschritte zu vereinfachen, Durchlaufzeiten von Prozessen zu verkürzen, Papier zu eliminieren oder die automatische Aufzeichnung von Qualitätsattributen einzuführen. Idealweise wird dazu die gegenwärtige Ist-Situation vor der Einführung der angedachten Lösung beschrieben sowie die angestrebte Soll-Situation nach Einführung der Lösung.
- Wie sieht der Nutzungskontext des zukünftigen Systems aus?
Ohne Verständnis des zugrundeliegenden Nutzungskontexts des zukünftigen Systems ist eine adäquate Systementwicklung kaum möglich. Geschäfts- und Teilprozesse (Abfolgen von Einzeltätigkeiten, die schrittweise manuell/automatisiert ausgeführt werden sollen, um ein fachliches Ziel zu erreichen) sind daher detailliert zu beschreiben, ggf. zu modellieren und die sich daraus ergebenden Szenarien oder Anwendungsfälle für das System zu skizzieren.
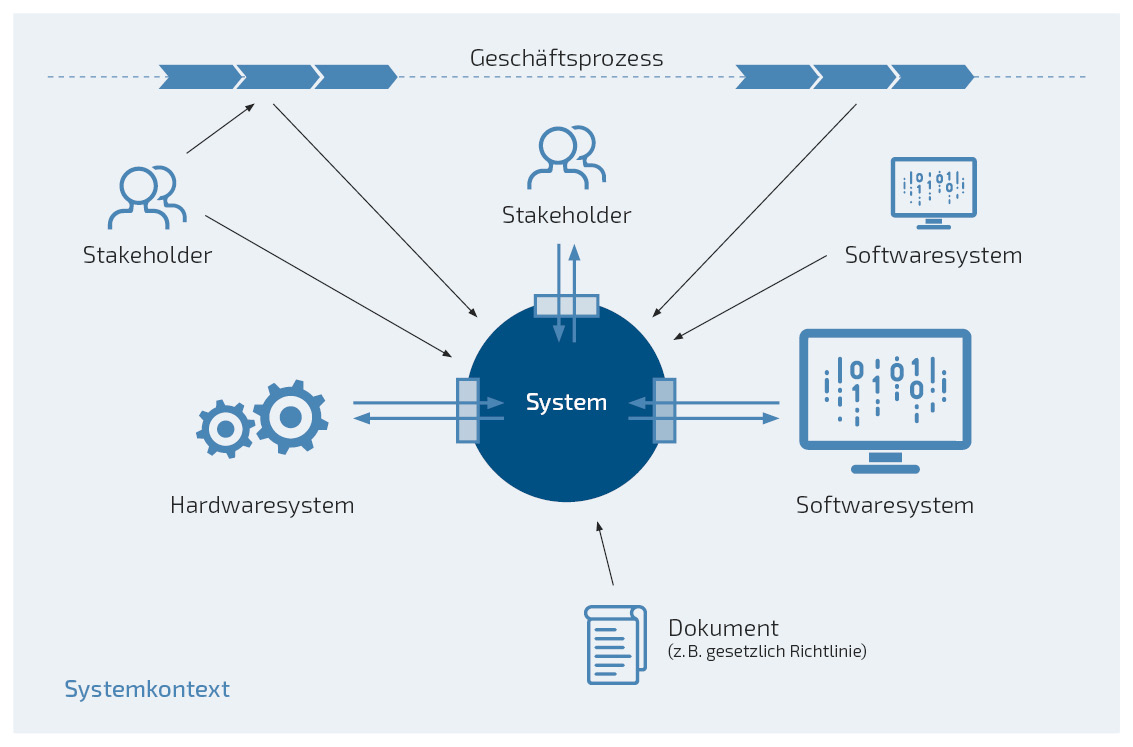
- Was gehört zum System und was zum Systemkontext?
Diese Frage sollte so früh wie möglich beantwortet werden, idealerweise vor der eigentlichen Anforderungserhebung, um zu entscheiden, wer und was Einfluss auf die Anforderungen an ein System hat (Anforderungsquellen) und um den Umfang des Systems bzw. der Systementwicklung zu kennen (In-Scope vs. Out-of-Scope). Nur mit einem vollständig definierten Systemkontext ist es möglich, die Anforderungen an das System vollständig zu erfassen und in die Systementwicklung einfließen zu lassen. Das System steht hier für das computergestützte System (z. B. ein Leitrechner mit Software(modulen), Hardwarekomponenten, Systemdokumentation), der Systemkontext für die relevante Umgebung (z. B. die Produktionsanlage, Mitarbeiter*innen, andere Softwaresysteme) und die Systemgrenze für die Schnittstellen dazwischen (z. B. Daten, die Mitarbeiter*innen in das System eingeben können sollen). Die Abgrenzung des Systemkontextes ist entscheidend für die Beantwortung drei wichtiger Fragen:
- Was soll entwickelt werden (gestalt- und veränderbar)?
- Was hat Einfluss auf die Entwicklung (nicht gestalt- und veränderbar, aber für Ermittlung der Systemanforderungen zu berücksichtigen)?
- Was kann (nach aktuellem Kenntnisstand) vernachlässigt werden?
Wer sind die Stakeholder des zukünftigen Systems, welche Bedürfnisse und welchen Einfluss auf die zu entwickelnde Lösung haben sie?
Hierbei sollten alle internen und externen Stakeholder berücksichtigt werden, die direkte oder indirekte Ansprüche oder Interessen an der zu entwickelnden Lösung (z.B. Produktionssteuerung) haben. Hierzu gehören zum Beispiel auch die Personen und -gruppen, die das zukünftige System betreiben, warten oder schulen sollen.
Wie kann Kontron AIS bei der Digitalisierung der Produktion unterstützen?
Als Entwickler und Integrator für Softwarelösungen zu Produktionssteuerung und Konnektivität im GMP-Umfeld, können Kunden von erprobten Produkten wie unserem FabEagle®LC Leitrechner oder unserer Low-Code-Schnittstellintegrationssoftware FabEagle®Connect profitieren. Sowohl unsere Produkte als auch die Projektentwicklung erfüllen die GMP-Anforderungen und basieren auf den GAMP-Richtlinien und stellen eine reibungslose Integration nach dem V-Model sicher. So bilden sie die Basis, von der aus auch vielfältige kunden- bzw. prozessspezifische Erweiterungen realisiert werden können.
Für eine nachhaltige Digitalisierung analoger Produktionsprozesse ist neben der passenden Software auch ein systematisches Requirements Engineering entscheidend. Wir unterstützen daher bei der Ermittlung, Dokumentation, Prüfung und Abstimmung der Kunden- und Systemanforderungen im Lasten- und Pflichtenheft sowie dem Anforderungsmanagement über den Lebenszyklus. In Workshops erarbeiten wir gemeinsam die spezifischen Anforderungen der Produktionsprozesse an die Produktionssteuerung und Konnektivität.
1 https://www.recht.bund.de/bgbl/1/2023/197/VO.html, 31.08.2023
2 https://www.bfarm.de/DE/Medizinprodukte/_node.html; 30.08.2023
Sie benötigen Unterstützung bei der Digitalisierung Ihrer Produktion entsprechend GMP-Anforderungen und GAMP-Richtlinien? Ich bin Ihr Ansprechpartner!
